In recent years, as competition in the generics market intensifies and R&D costs for innovative drugs continue to rise, modified new drugs have emerged as a crucial development direction for the global pharmaceutical industry. The United States has accumulated substantial experience through its 505(b)(2) pathway, while China has witnessed rapid growth in applications and approvals since establishing clear registration categories for modified new drugs in 2016. This article integrates successful practices from the United States 505(b)(2) pathway to analyze China's policies and regulations, acceptance/approval status, and R&D Status for modified new drugs, while exploring existing challenges and future optimization direction.
1. Overview and Market Experience of the United States 505(b)(2) Pathway
1.1 Path definition and classification
The United States 505(b)(2) pathway, originating from the 1984 Hatch-Waxman Act, aims to provide a simplified approval process for products that involve modifications to the dosage form, formulation, indications, or other aspects of already marketed drug active ingredients. According to FDA classifications, it covers 10 categories of registration applications including new dosage forms (Category 3), new combinations (Category 4), and new indications (Category 6).
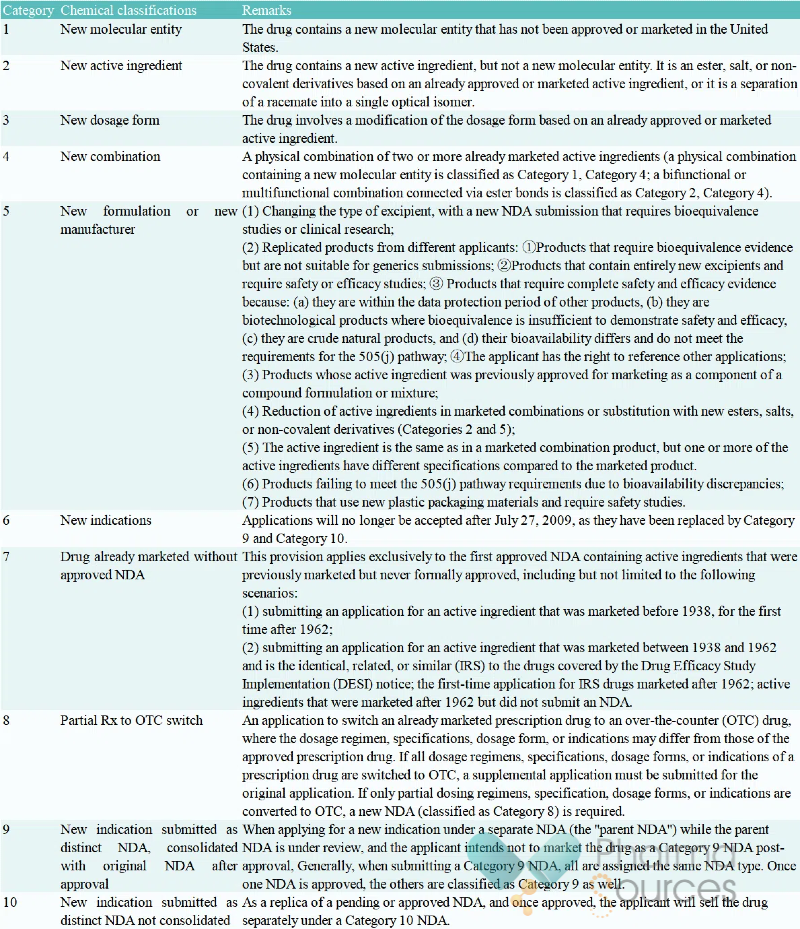
Source: Manual of Policies and Procedures
The United States New Drug Application (NDA) Registration Classifications for Pharmaceutical Chemicals (Image Source: China Food and Drug Administration)
1.2 Market performance
Market Scale: As of 2023, over 2,500 products under the 505(b)(2) pathway have been approved in the United States cumulatively, with 2022 sales volume reaching USD 105 billion, accounting for 17% of the pharmaceutical market.
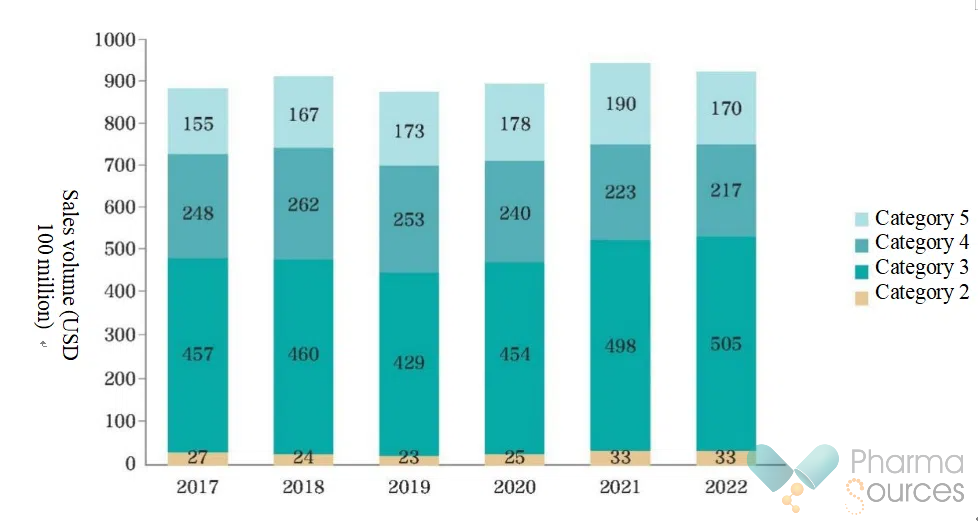
Market Sales Volume for United States Categories 2-5 505(b)(2) Products from 2017 To 2022 (Source: China Food and Drug Administration)
1.3 Success factors
Creative design: Restasis addresses dry eye disease through immunomodulatory mechanisms.
Technological barriers and patent protections: Complex preparations (such as leuprorelin microspheres, octreotide microspheres) have a long lifecycle due to the high difficulty in generic formulation.
Clinical advantages: Such as liposomes doxorubicin reducing cardiotoxicity and albumin-bound paclitaxel enhancing efficacy.
Precise market positioning: Originator enterprises extend the product lifecycle through brand extensions, such as the risperidone orally disintegrating tablet.
1.4 Review characteristics and challenges
Efficiency of review: An average review cycle is of 18.4 months, with 30% of approvals relying on clinical pharmacology data.
Market concentration: The top 50 products constituting 72% of total sales volume, dominated by originator enterprises, while small and medium enterprises face promotion and healthcare access pressures.
2. China's modified new drugs policies and regulations, regulations and registration classification
2.1 Policy evolution
In 2015, the country launched a drug approval reform, and in 2016, the Pharmaceutical Chemicals Registration Classification Reform Work Plan first clarified the definition of modified new drugs (Category 2), requiring that they "demonstrate clear clinical advantages". The 2020 Drug Registration Management Measures refined accelerated pathways including priority review and conditional approval.
2.2 Registration categories and requirements
Category 2.1: New active ingredients (such as isomers or new salts) must demonstrate clinical advantages.
Category 2.2: New dosage forms and new formulation processes must be validated through bioequivalence or clinical trials.
Category 2.3: New combination formulations emphasize the synergistic effects of the components.
Category 2.4: New indications must be supported by independent clinical trials.
Available data reveal that Category 2.2 (new dosage forms) and Category 2.4 (new indications) dominate over 90% of China's modified new drugs, yet face issues like homogeneous competition and high R&D barriers. Between 2016 and 2021, for pharmaceutical chemicals modified new drugs, whether applying for clinical trials or production, the majority of the registration classifications were concentrated in Category 2.2 and 2.4. The numbers for Category 2.1 and 2.3 were relatively small, and in some cases, there were none.
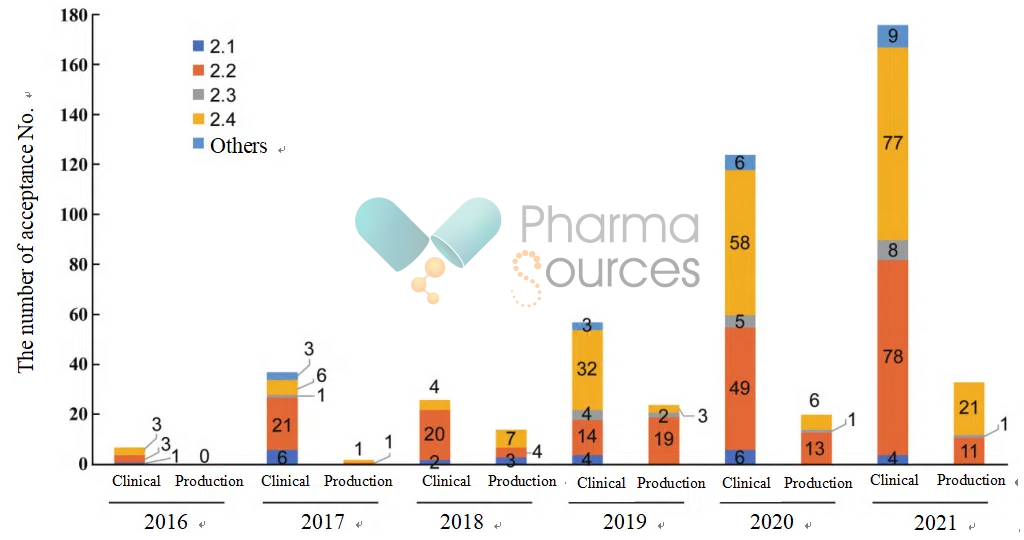
The 2016-2021 Application Status of Modified New Drugs of Pharmaceutical Chemicals (Source: China Journal of New Drugs)
2.3 Review/approval characteristics (2016-2023)
Application trends: There were 1,496 clinical trial applications (IND), 278 production applications (NDA), and 165 approvals.
Category distribution: Category 2.4 (new indications) accounted for 62%, Category 2.2 (new dosage forms) 29%, with Categories 2.1 and 2.3 combined totaling less than 10%.
Therapy field: Anti-tumor drugs (38%), nervous system drugs (18%), and anti-infective drugs (12%) are the key areas of R&D focus.
Dosage form innovations: Sustained and controlled release formulations (19.7%) and sterile powder for injection (16.4%) dominated Category 2.2 developments.
3. The Comparison between China and the United States: Similarities and Differences in Policies and Markets
3.1 Policy differences
Clinical advantage requirements: China emphasizes "significant clinical advantages" as a prerequisite for approval, while the United States prioritizes market validation.
Data protection: The United States provides a 3-year data exclusivity period for products containing clinical trials, while China does not yet have clear regulations on this matter.
Review flexibility: The United States establishes effective communication with the FDA through the Pre-IND meeting before submitting a new drug clinical trial application, in order to seek exemptions or reductions in clinical trial requirements. In comparison, China's review standards are relatively strict.
3.2 Market performance differences
Type of enterprise: In the United States, the original innovator enterprises lead the market, while in China, generics pharmaceutical companies are primarily focusing on transformation.
Product type: New dosage forms (Category 3) account for a larger proportion in the United States, while in China, new indications (Category 2.4) dominate.
Competitive landscape: In China, there is a phenomenon of "concentrated submissions" (e.g., paclitaxel micelles), while the United States market is more concentrated.
4. R&D Status and Challenges of Modified New Drugs in China
4.1 Status analysis
Policy-driven focus: Anti-tumor and neurological drugs align with the "Healthy China" strategic direction.
Insufficient technological accumulation: The proportion of high-end preparations (such as liposomes and microspheres) is low, with a reliance on imported technologies.
Homogeneous competition: In 2021, multiple enterprises focus on submitting applications for butylphthalide injection, which led to delays in the approval process.
4.2 Core challenges
High R&D barriers: Category 2.1 drugs (new salts/isomers) require large-scale clinical trial that strain small-to-medium enterprises.
Uncertain market returns Some products have no significant clinical advantages, making it difficult to overcome the pressure of generics substitution.
Weak patent portfolios: Patents for dosage modifications are easily circumvented, with insufficient technological barriers.
5. Lessons from the United States Experience for China
The United States experience demonstrates that emphasizing clinical advantages, focusing on patent protection, and market positioning are key to success. Specifically, the lessons from the United States experience for China can be summarized in the following aspects:
5.1 Optimizing R&D strategies
Focus on differentiated innovation: Avoid simple dosage improvements and explore combination therapies (e.g., dextromethorphan + quinidine) and new drug delivery methods (e.g., transdermal patches).
Strengthen patent protection: Focus on preparation processes and use patents to extend market exclusivity period.
5.2 Policy suggestions
Improve data protection systems: granting data exclusivity periods for modified new drugs containing clinical trials;
Enhancing review flexibility: Through establishing Pre-IND communication mechanisms to permit partial test waivers;
Guide resource allocation: Encourage the development of drugs for shortage fields, such as anti-infective and pediatric medications, through priority review.
5.3 Enterprise action direction
Collaborate with originator companies: Adopting United States models, enabling small and medium enterprises to partner with innovators for upgraded product development;
Clinical demand-driven guiding: Designing products focused on patient adherence (e.g., orally disintegrating tablets) and safety (e.g., liposomes).
Consequently, China must further refine policy guidance to incentivize differentiated innovation, elevate technological barriers, and achieve high-quality development of modified new drugs.
6. Conclusion
Overall, modified new drugs serve as a critical bridge between generics and innovative drugs. Over four decades, the United States 505(b)(2) pathway has cultivated a clinical value-centric market ecosystem; While China started later, its policy dividends have driven rapid growth in application volumes. In the future, China needs to further balance regulatory rigor with review flexibility, encouraging companies to shift from "modifying for the sake of modification" to "modifying based on needs." At the same time, strengthening international cooperation and technology transfer is expected to further drive modified new drugs to become a new growth engine for pharmaceutical innovation.
References:
1. Center for Drug Evaluation, NMPA, https://www.cde.org.cn/
2. U.S. Food and Drug Administration, https://www.fda.gov/
https://www.nmpa.gov.cn/xxgk/ggtg/ypggtg/ypqtggtg/20160309151801706.html
3. Announcement No. 51 of 2016 by the NMPA: Work Plan for Reform of Pharmaceutical Chemicals Registration Classifications, https://www.nmpa.gov.cn/xxgk/ggtg/ypggtg/ypqtggtg/20160309151801706.html
4. Sah B, Kumar S. Regulatory Foundation of 505(B)(2) Application in US FDA[J]. IJMAR. 2023,11(4):1-7.
5. Salminen WF, Aloba O, Drew A, Marcinowicz A, Huang M. US FDA 505(b)(2) NDA clinical, CMC and regulatory strategy concepts to expedite drug development. Drug Discov Today. 2023, 28(7):103618.
About the author:
Kevin, engaged in research on clinical drug evaluation and rational medication use, as well as pharmaceutical affairs management.




 ALL
ALL Pharma in China
Pharma in China Pharma Experts
Pharma Experts Market News
Market News Products Guide
Products Guide Brand Story
Brand Story

































 Pharma Sources Insight January 2025
Pharma Sources Insight January 2025








