As of Wednesday 26 May 2021, Europe’s Medical Device Regulation (MDR) (Regulation (EU) 2017/745 on medical devices) is applicable in the European Union.
The MDR, which was adopted in April 2017, changes the European legal framework for medical devices and introduces new principal and supportive responsibilities for the European Medicines Agency (EMA) and for national competent authorities in the assessment of certain categories of products.
The Regulation entered into force in May 2017 and had a staggered transitional period.
The MDR introduces new or revised responsibilities for EMA for:
medicines with an integral device, such as pre-filled syringes and pens, and pre-filled inhalers;
medical devices containing an ancillary medicinal substance to support the proper functioning of the device. Examples include drug-eluting stents, bone cement containing an antibiotic, catheters coated with heparin or an antibiotic agent and condoms coated with spermicides;
medical devices made from substances that are absorbed by the human body to achieve their intended purpose;
borderline products for which there is uncertainty over which regulatory framework applies. Common borderlines are between medicinal products, medical devices, cosmetics, biocidal products, herbal medicines and food supplements.
Further details are available on the Medical devices page of the EMA’s website..
To support the implementation of the MDR, the EMA stated that it is currently preparing and will soon publish updated guidance on quality requirements for medical devices in human medicines that include a medical device, as well as an updated Q&A.
The MDR replaces the existing Directives for medical devices (93/42/EEC and 90/385/EEC). The Regulation on in vitro diagnostic medical devices, which also came into force in May 2017, will replace Directive 98/79/EC when it comes into application on 26 May 2022.




 ALL
ALL Pharma in China
Pharma in China Pharma Experts
Pharma Experts Market News
Market News Products Guide
Products Guide Brand Story
Brand Story


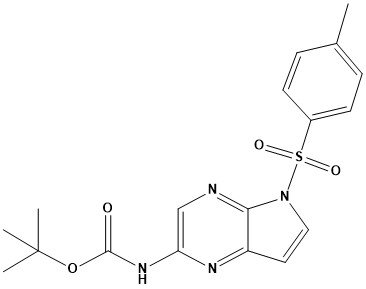
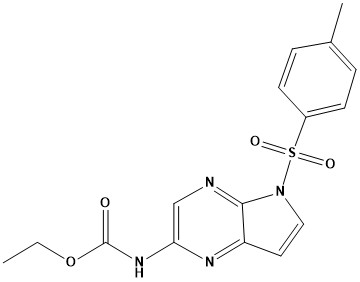
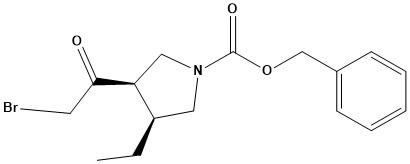
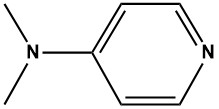

























 Pharma Sources Insight January 2025
Pharma Sources Insight January 2025








