The U.S. Food and Drug Administration (FDA) posted the seventh spreadsheet of reportable adverse events that Bayer is required to submit as outlined in the April 24, 2020 variance from MDR reporting requirements. As stated there, the FDA requires Bayer to process all reportable adverse events it becomes aware of in social media in connection with litigation within one year of the date of the variance, unless an extension is granted. The variance is limited to MDR-reportable events for Essure that Bayer is or becomes aware of for the period between November 2016 through November 2020.
As this information is based on social media posts, these reports may reference information already reported to the FDA and do not necessarily represent new adverse events. The FDA will continue to make the adverse event information submitted under the variance publicly available on this web page after spreadsheets are received from Bayer and reviewed by the FDA. Summary information about the events in each spreadsheet is also accessible in the FDA’s MAUDE database.
Bayer posted the second quarterly analysis report of the adverse event information included in the fourth, fifth, and sixth variance reporting spreadsheets for the reporting time period of September 2020 through November 2020. The second quarterly analysis report includes a total of 11,884 reportable events with the following report type: 11,830 serious injury reports, 45 malfunction reports, and 9 death reports. Reports do not necessarily represent unique cases, but rather events identified in comment threads from social media posts, sometimes by the same individual. The most common patient problems reported were pain, perforation, foreign body/device fragment in patient, pregnancy (including ectopic), heavier periods (menses/hemorrhage), and hypersensitivity. The majority of reports (94%) are related to potential device removal which is consistent with the MDRs for Essure since 2017. For the 9 death reports, the social media posting dates (when provided) ranged from the year 2013 to 2015. None of the death reports provided a date for the reported death event. Based on the limited information in the event descriptions for the reports and the nature of the information, it is difficult to identify duplicate reports within the spreadsheet of events, as well as duplicate reports previously submitted to the FDA. The limited information prevents the ability to draw any conclusions as to whether the device, or its removal, caused or contributed to any of the reported deaths or other events in the reports.
Bayer will continue to provide quarterly analysis reports and a final analysis report of the adverse event information submitted under the variance, which Bayer will make publicly available.
The FDA relies on adverse event and product problem reports, FDA-mandated postmarket studies, and published literature to monitor the safety and effectiveness of medical devices. The FDA reviews the available information about Essure and the experiences of patients who have or had Essure implants. This includes experiences of patients who had positive outcomes with Essure as well as those who experienced problems.
Even though Essure is no longer being sold or distributed in the United States, the FDA will continue to evaluate medical device reports related to Essure and will keep the public informed if new information becomes available.
Adverse event and product problem reports submitted to the FDA are one source the FDA uses to monitor the safety profile of medical devices. These reports may contribute to the detection of potential device-related safety issues as well as to the benefit-risk assessments of these devices. While such reports are a valuable source of information, this type of reporting system has notable limitations, including the potential submission of incomplete, inaccurate, untimely, unverified, or biased data. This can make it difficult for the FDA to confirm whether a device caused a specific event based only on the information provided in a medical device report. Complaints or adverse event reports do not necessarily directly indicate a faulty or defective medical device, and adverse event reports alone cannot be used to establish or compare rates of event occurrence. Additionally, the FDA may receive multiple reports related to the same event, making it difficult to determine actual numbers of events.




 ALL
ALL Pharma in China
Pharma in China Pharma Experts
Pharma Experts Market News
Market News Products Guide
Products Guide Brand Story
Brand Story


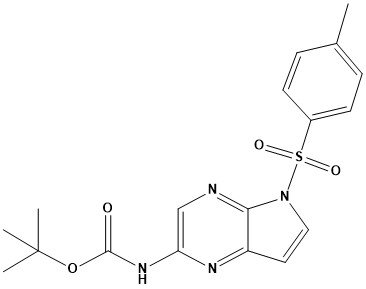
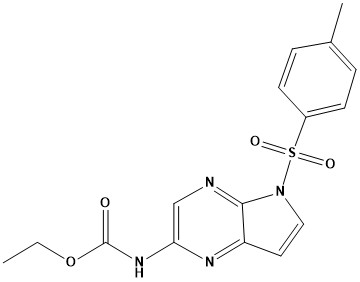
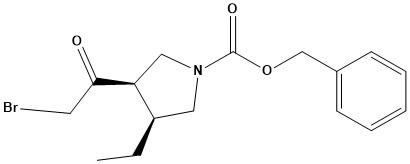
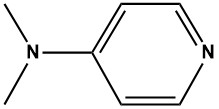

























 Pharma Sources Insight January 2025
Pharma Sources Insight January 2025








