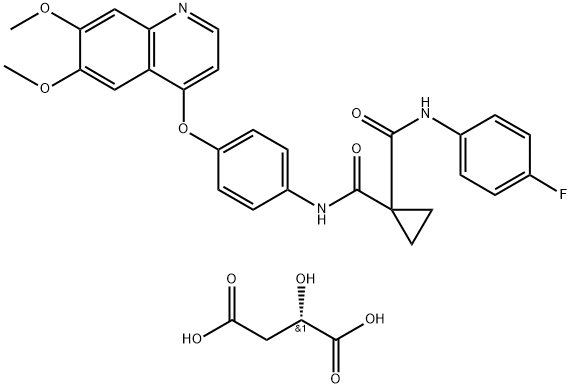
Cabozantinib, a tyrosine kinase inhibitor, was shown to reduce tumour volume and pain in patients with the genetic disorder neurofibromatosis type 1 (NF1). Because of the promising response rate, the Neurofibromatosis Clinical Trials Consortium has opened and fully enrolled a paediatric cohort (patients ages 3 to 15).
NF1 is a rare tumour predisposition syndrome which affects approximately 1 in 3000 people worldwide. The condition involves the proliferation of tumours throughout the central and peripheral nervous system. One of the most prevalent type of tumours in NF1 are plexiform neurofibromas (PN), multicellular tumours composed of tumorigenic Schwann cells, fibroblasts, perineural cells, macrophages, mast cells and secreted collagen. These PN tumors arise within nerves and affect almost 50 percent of patients with NF1. They grow rapidly during childhood and can lead to motor and sensory dysfunction, pain and disfigurement. When the tumors impinge on vital structures like the airway or the spinal cord, they can be life-threatening. While they are benign, PN tumours can become malignant over time.
Surgery is the current standard of care because chemo- and radiotherapies are ineffective. However, given that the tumours can be intertwined with nerves and other vital structures, surgery is often not possible. Recent studies have shown that a MEK inhibitor called selumetinib can be an effective treatment in some children with NF1-related PNs, but not all patients respond to this treatment, so there is a need for more treatment options.
Based on preclinical studies of cabozantinib, an investigational drug that targets both the tumorigenic Schwann cells in PNs as well as the complex tumor microenvironment, researchers at Children’s Hospital of Philadelphia (CHOP), US, enrolled patients in a single arm, multicentre Phase II clinical trial. The results of the trial were published in Nature Medicine.
Twenty-three patients aged 16 to 34 enrolled in the trial; twenty-one were evaluable for drug toxicity and 19 were evaluable for their response to the treatment. Of the 19 patients studied for response, eight (42 percent) had a greater than a 20 percent decrease in tumour volume (partial response) and 11 had stable disease after 12 rounds of treatment. No patient had disease progression while participating in the trial. Those with a partial response to cabozantinib also reported a significant reduction in tumour pain intensity and pain interference in daily life.
The adverse events reported by participants included diarrhoea, nausea, asymptomatic hypothyroidism, fatigue and palmar plantar erythrodysesthesia, a condition that causes redness, swelling and pain on the palms of the hands and/or the soles of the feet. None of these effects were reported as being severe.
“This is the second class of drugs to demonstrate a very promising response rate for NF1 patients with these tumours,” said first author Dr Michael Fisher, Chief of the Section of Neuro-Oncology and Director of the Neurofibromatosis Program at CHOP, and Group Chair for the NF Clinical Trials Consortium, which includes 25 sites developing innovative biologically-based clinical trials for complications of NF. “Collectively, the data presented in this study illustrate a true bench-to-bedside approach, coordinating translational and clinical efforts to advance targeted therapies for a rare disease like NF1.”
He added: “It’s incredibly exciting that we now have two classes of drugs that result in tumor responses, given that we had no promising agents only a few years ago. However, despite this excitement, neither cabozantinib nor MEK inhibitors shrink all tumours or make them go away completely. Therefore, we are building on these results as well as ongoing laboratory studies and are planning future exploration of combination therapies, so that we can further improve outcomes for these patients with these debilitating and life-threatening tumours.”




 ALL
ALL Pharma in China
Pharma in China Pharma Experts
Pharma Experts Market News
Market News Products Guide
Products Guide Brand Story
Brand Story



























 Pharma Sources Insight January 2025
Pharma Sources Insight January 2025








