Neurogene, a company founded to bring life-changing genetic medicines to patients and families affected by rare neurological diseases, announces that the European Medicines Agency (EMA) has granted orphan drug designation (ODD) to its adeno-associated virus vector (AAV) with engineered transgene encoding the human AGA gene for patients diagnosed with aspartylglucosaminuria.
Aspartylglucosaminuria (AGU) is an inherited rare, progressively debilitating neurodegenerative lysosomal storage disorder. Currently there are no approved treatments to slow or cure AGU in patients living with this disease.
“Due to the lack of approved treatment options and the severity of symptoms, the prognosis and quality of life for patients and families affected by AGU can often be very poor,” said Minna Laine, M.D., Ph.D., Child Neurologist, Helsinki University Hospital, Helsinki, Finland. “The EMA granting Orphan Drug Designation for a potential gene therapy for AGU represents promise while underscoring the urgent, unmet medical need for patients around the world diagnosed with this condition.”
The AGU Orphan Drug Designation represents Neurogene’s third ODD for a rare neurological disease. Earlier this year the U.S. Food and Drug Administration (FDA) granted Neurogene ODD for gene therapies addressing CLN5 and CLN7, two subtypes of Batten disease which are also neurodegenerative and have no available treatment options to slow or halt the disease.
“Gaining this important regulatory milestone designation means we are one step closer to moving the first AGU treatment option into the clinic,” said Rachel McMinn, Ph.D., Neurogene’s Founder and Chief Executive Officer. “Neurogene is collaborating with regulatory authorities, families and patients to make a safe and effective gene therapy for AGU available as quickly as possible.”
EMA orphan designation is designed to encourage the development of new treatments for life-threatening or chronically debilitating conditions that are rare (affecting not more than five in 10,000 people in the European Union). Medicines that meet the EMA’s orphan designation criteria qualify for several incentives, including 10 years of market exclusivity, protocol assistance, and potentially reduced fees for regulatory activities. Applications for orphan designation are examined by the EMA’s Committee for Orphan Medicinal Products (COMP), using the Committee’s network of experts.
Aspartylglucosaminuria (AGU) is an inherited rare, progressively debilitating neurodegenerative lysosomal storage disorder caused by a variant in the AGA gene, which causes deficient activity in the aspartylglucosaminidase (AGA) enzyme and leads to the accumulation of toxic material in the brain and other organ systems. The hallmark symptom of AGU is developmental delay and affected individuals over time experience a constellation of signs and symptoms that may include chronic ear and respiratory infections, progressive speech delay, gait disturbance, gastrointestinal disturbances, behavioral problems, and cognitive defects. Seizures may develop during late adolescence and continue throughout adulthood. Ultimately, AGU leads to erosion of basic daily living skills and is associated with premature death. AGU can be confused with other more common neurological diseases, leading to a delayed or incorrect, diagnosis.
Neurogene is committed to lowering the barriers of obtaining a genetic diagnosis for patients and has partnered with Invitae to co-sponsor two genetic testing programs in the US and in Canada. Healthcare providers can order, at no cost, an Invitae Epilepsy panel for any child under the age of eight who has had an unprovoked seizure. The Detect Lysosomal Storage Diseases panel is available to patients at no charge who are suspected of having a lysosomal storage disease.




 ALL
ALL Pharma in China
Pharma in China Pharma Experts
Pharma Experts Market News
Market News Products Guide
Products Guide Brand Story
Brand Story


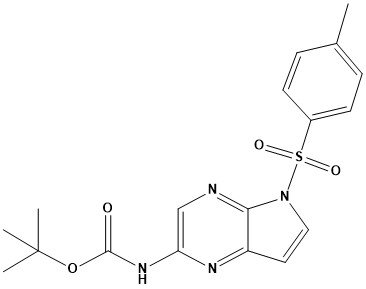
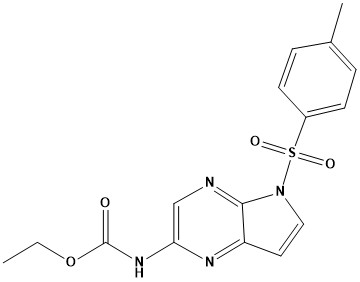
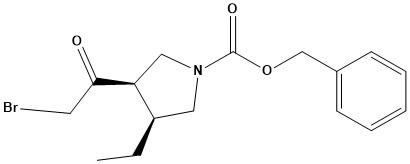
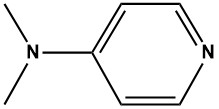


























 Pharma Sources Insight January 2025
Pharma Sources Insight January 2025








