The US Food and Drug Administration is extending the enforcement discretion policy for certain human cell, tissue, and cellular and tissue-based products (HCT/Ps). As a result of the challenges presented by the COVID-19 public health emergency, the FDA is extending its enforcement discretion policy which will provide manufacturers and potential sponsors an additional six months to determine if they need to submit an Investigational New Drug (IND) or marketing application and, if such an application is needed, to prepare and submit the application, as appropriate.
The FDA’s November 2017 regenerative medicine policy framework was developed to help facilitate and support the innovation of regenerative medicine therapies. The agency also outlined its intent to exercise enforcement discretion, until November 2020, for certain regenerative medicine products, with respect to the FDA’s IND and premarket approval requirements; to give manufacturers time to determine what requirements apply as well as engage with the agency. The US FDA is revising the guidance to reflect that FDA generally intends to exercise enforcement discretion with respect to IND and premarket approval requirements for certain HCT/Ps through May 2021.
“To be clear, our policy of enforcement discretion only pertains to certain human cell, tissue and cellular and tissue-based products that do not raise potential significant safety concerns or reported safety concerns. This policy was never intended to provide a cover for bad actors, and we intend to continue to take action against manufacturers and health care providers who are offering unapproved regenerative medicine products that have the potential to put patients at significant risk,” said Peter Marks, M.D., Ph.D., director of the FDA’s Center for Biologics Evaluation and Research (CBER).
Since the issuance of the comprehensive regenerative medicine policy framework in late 2017, the FDA has worked with product developers to determine if they need marketing authorization application and, if so, how they should submit their application to the FDA.
Importantly, the agency’s enforcement discretion policy for IND and premarket approval requirements does not apply to products that have been associated with reported safety concerns or have the potential to cause significant safety concerns to patients. The agency has stepped up its oversight of cellular and related products in recent years and has issued compliance actions, including numerous warning and untitled letters, and pursued litigation for serious violations of the law, including some involving patient harm.
Patients should make sure that any regenerative medicine product they are considering is either FDA-approved or being studied under an IND, which is a clinical investigation plan submitted and allowed to proceed by the FDA.
The FDA, an agency within the U.S. Department of Health and Human Services, protects the public health by assuring the safety, effectiveness, and security of human and veterinary drugs, vaccines and other biological products for human use, and medical devices. The agency also is responsible for the safety and security of our nation’s food supply, cosmetics, dietary supplements, products that give off electronic radiation, and for regulating tobacco products.




 ALL
ALL Pharma in China
Pharma in China Pharma Experts
Pharma Experts Market News
Market News Products Guide
Products Guide Brand Story
Brand Story


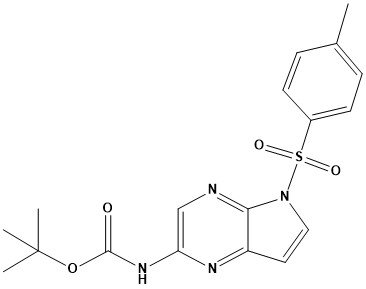
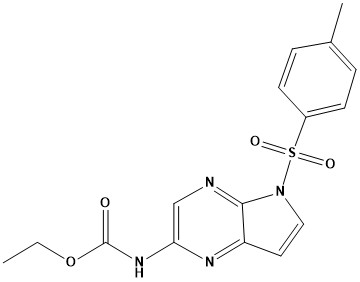
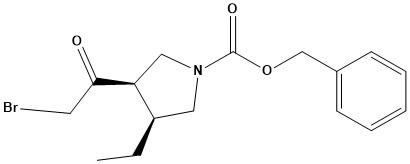
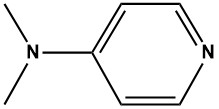
















![(E)-2-(6,7-dihydro-1H-indeno[5,4-b]furan-8(2H)-ylidene)ethanamine](https://eimg.pharmasources.com/image/product/20231221/b3d4562fe556876869275b2eabd2660ajpg)

![11,12-Dihydroindolo[2,3-a]Carbazole](https://eimg.pharmasources.com/image/product/20230504/7d8a7792b76939f79c62660808de627fjpg)





 Pharma Sources Insight January 2025
Pharma Sources Insight January 2025








