The Food and Drug Administration (FDA) approved pembrolizumab (KEYTRUDA, Merck) for the first-line treatment of patients with unresectable or metastatic microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) colorectal cancer.
Approval was based on KEYNOTE‑177 (NCT02563002), a multicenter, international, open-label, active-controlled, randomized trial that enrolled 307 patients with previously untreated unresectable or metastatic MSI-H or dMMR colorectal cancer. Determination of MSI or MMR tumor status was made locally using polymerase chain reaction (PCR) or immunohistochemistry (IHC), respectively. Patients were randomized (1:1) to receive pembrolizumab 200 mg intravenously every 3 weeks or investigator’s choice of mFOLFOX6/FOLFIRI ± bevacizumab or cetuximab given intravenously every 2 weeks. Patients randomized to chemotherapy were offered pembrolizumab at the time of disease progression.
The main efficacy outcome measures were progression-free survival (PFS) and overall survival (OS). Median PFS was 16.5 months (95% Confidence Interval [CI]: 5.4, 32.4) in the pembrolizumab arm and 8.2 months (95% CI: 6.1, 10.2) in the chemotherapy arm (HR 0.60, 95% CI 0.45, 0.80; two-sided p-value=0.0004). At the time of the PFS analysis, the OS data were not mature.
The most common adverse reactions reported in ≥20% of patients receiving pembrolizumab as a single agent are fatigue, musculoskeletal pain, decreased appetite, pruritus, diarrhea, nausea, rash, pyrexia, cough, dyspnea, constipation, pain, and abdominal pain.
The recommended pembrolizumab dose for MSI-H/dMMR colorectal cancer is 200 mg every 3 weeks or 400 mg every 6 weeks.
This review was conducted under Project Orbis, an initiative of the FDA Oncology Center of Excellence. Project Orbis provides a framework for concurrent submission and review of oncology drugs among international partners. For this application, a modified Project Orbis was undertaken because of the timing of submission to other regulatory agencies. FDA is collaborating with the Australian Therapeutic Goods Administration, Health Canada, and Swissmedic. The review is ongoing for these three agencies.
This review used the Real-Time Oncology Review (RTOR) pilot program, the Assessment Aid, a voluntary submission from the applicant to facilitate the FDA’s assessment, and Summary Level Review, which relied on qualified data summaries to support approval of a supplemental application. The FDA approved this application approximately 5 months ahead of the FDA goal date.




 ALL
ALL Pharma in China
Pharma in China Pharma Experts
Pharma Experts Market News
Market News Products Guide
Products Guide Brand Story
Brand Story
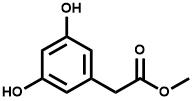
![2-[[(3aR,4S,6R,6aS)-6-amino-2,2-dimethyl-tetrahydro-3aH-cyclopenta[d][1,3]-dioxol-4-yl]oxy]-1-ethano](https://eimg.pharmasources.com/image/product/20250409/9cc6a4e71d673030f801f638c61fdd84png)




























 Pharma Sources Insight January 2025
Pharma Sources Insight January 2025








