The Food and Drug Administration (FDA) is a federal agency of the United States Department of Health and Human Services, that regulates the use of drugs within the United States. As per FDA mission statement, “the Food and Drug Administration is responsible for protecting the public health by ensuring the safety, efficacy, and security of human and veterinary drugs, biological products, and medical devices; and by ensuring the safety of our nation’s food supply, cosmetics, and products that emit radiation.” Its primary responsibility is ensuring the protection of human subjects who participate in clinical studies testing new drugs such as Investigational New Drug (INDs).
Patient safety evaluation during clinical studies is a critical component in all stages of the drug development life-cycle. Pharmaceutical sponsors need to adequately characterize the safety profile of the drug in order to obtain regulatory approval and marketing authorization.
IND Safety Reports
In the US, prior to the initiation of a first in human clinical trial, pharmaceutical sponsors must submit an Investigational New Drug (IND) application to the FDA as required by law. The FDA reviews the IND (typically within 30 calendar days) for safety to ensure that research subjects will not be subjected to unreasonable risk.
Under 21 of the code of federal regulations (CFR) 312.32(c), the sponsor is required to notify FDA and all participating investigators in an IND safety report (i.e., 7-or15-day expedited report) of serious and unexpected suspected adverse reactions from clinical trials or any other source as soon as possible, but no later than 15 calendar days after the sponsor receives the safety information and determines that the information qualifies for reporting. If applicable, relevant follow-up information to an initial safety report must be submitted as a Follow-up Safety Report as well.
Any adverse event associated with the use of an IND drug in humans should be reported as soon as possible, whether or not considered drug related, including:
- Use in professional practice
- Overdose (intentional and accidental)
- Abuse
- Withdrawal
- Failure of expected pharmacological action (lack of effect)
Electronic Submission Of IND Safety Reports To FAERS
According to the FDA, all sponsors who conduct clinical trials under an IND are required to submit IND safety reports as per 21 CFR 312.32 for suspected adverse reactions (AE) that are observed in the clinical study, at domestic or foreign study sites.
These reports have traditionally been submitted to FDA on MedWatch Form FDA 3500A or on a Council for International Organizations of Medical Sciences (CIOMS) I Form or other narrative forms. Currently these IND safety reports for sponsors of commercial INDs are submitted in eCTD format (electronic common technical document) generally as PDFs, FDA takes this data forms and converts into the International Conference on Harmonisation (ICH) E2B (an international standard for transmitting medicine adverse event reports) to FDA Adverse Event Reporting System (FAERS), which is a very inefficient and labor-intensive process to both tracking review, and regulatory agencies. It is also lack of universal tracking system.
To address these problems, in October 2019, the FDA is establishing a new submission process for investigational new drug application (IND) safety reports when they electronically submit to the FDA FAERS, focusing particularly on this important safety information as structured data. FDA takes this data forms and convert into E2B to FAERS, then further develop some of the technical specifications around the particular data elements in E2B that are required for IND safety reports for the post-market. The new submission process will allow for use of data visualization and analytic tools for review and tracking.
It is also leverages existing processes that are used in the post-market safety reporting such as the ICH E2B data standard and also FDA submission pathway such as the gateway and the safety reporting portal. It also complies with existing FDA Federal Regulations surrounding IND safety reports on 21 CFR 312.32.
The new guidance will improve FDA’s ability to review and track safety signals that occur during the conduct of clinical trials. It will also provide sponsors with a reporting format which is consistent with ICH data standards and reporting to other regulatory agencies for serious and unexpected suspected adverse reactions that are required under 21 CFR 21 CFR 312.32(c)(1)(i).
What Is FDA Adverse Event Reporting System (FAERS)?
The FDA Adverse Event Reporting System (FAERS) is a database that contains information on adverse event and medication error reports submitted to FDA. The database is designed to support the FDA's post-marketing safety surveillance program for drug and therapeutic biologic products. The informatic structure of the FAERS database adheres to the international safety reporting guidance issued by the ICH E2B. Adverse events and medication errors are coded to terms in the Medical Dictionary for Regulatory Activities (MedDRA) terminology. FDA staff in CDER and CBER will regularly examine the FAERS database as part of routine safety monitoring. When a safety signal is identified from FAERS data, it will be further evaluated.
The new process will have sponsors submitting these IND safety reports as XML files to FAERS using the ICH E2B data standard, and this will allow FDA to receive standard data elements and be able to use data visualization and analytics tools for review and tracking purpose.
What Is XML file?
XML is a file extension for an Extensible Markup Language (XML) file format used to create common information formats and share both the format and the data on the World Wide Web, intranets, and elsewhere using standard ASCII text that can be read by both humans and machines. The structure of XML is based on a grouping of sections and elements that are annotated by start and end tags. XML is similar to HTML.
What Is ICH E2B Data Standard?
E2B does not have exact direct translation. According to ICH guidelines that E stands for efficacy. All E2 guidelines are associated with pharmacovigilance (drug safety). The ICH has defined E2B as the international standard for transmitting medicine adverse event reports. The ICH E2B document includes message standards required for effective transmission of individual case safety reports (ICSR). Eventually, the need for the exchange of high volume of safety information worldwide efficiently and automatically has led to periodic revisions of the E2B document.
Specifications For Submission Of IND Safety Reports To FAERS
The Electronic Common Technical Document (eCTD) is the standard format for submitting applications, amendments, supplements, and reports to FDA’s Center for Drug Evaluation and Research (CDER) and Center for Biologics Evaluation and Research (CBER). The new submission process of IND safety reporting will be required change in format for sponsors of commercial IND's under 745A (a) of the Federal Food, Drug, and Cosmetic Act (FD&C Act) which also applies to all subsequent submissions, including amendments, supplements, and reports, to the submission types identified.
Sponsors must submit IND safety reports to FAERS using one of the following options:
1 The Electronic Submission Gateway (ESG):
which is the FDA’s agency-wide solution for accepting electronic regulatory submissions. The FDA ESG enables the secure submission of premarket and post-market regulatory information for review. The FDA ESG is the central transmission point for sending information electronically to the FDA. XML files through the Gateway will be processed and stored in FAERS.
Or
2 The Safety Reporting Portal (SRP):
which streamlines the process of reporting product safety issues to the FDA and the National Institutes of Health (NIH).
Timetables And Requirements
The requirement to submit IND safety reports as described in this guidance will become effective 24 months after the publication of the final guidance. FDA will begin a voluntary submission process and will encourage sponsors of all INDs participating in the voluntary submission process once that is announced.
File Format Type For The eCTD Submissions
In general, documents should be provided in PDF searchable format. Images and other document types should be rendered into PDF format and retain searchable text whenever possible.
This provides specifications for submitting file format types using eCTD submission. It is a list of accepted file types and the eCTD locations in which those file types should be provided.
Acceptable File Formats For Use In eCTD
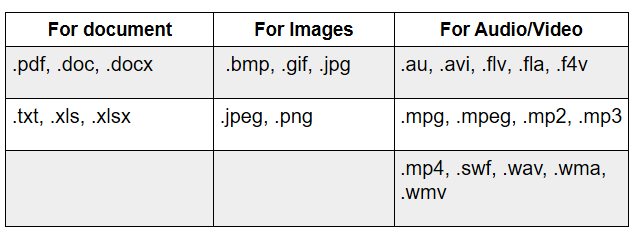
Type Of IND Safety Report And Key Data Elements Required
What to submit to FAERS:
- A single occurrence of an event that is uncommon and known to be strongly associated with drug exposure as per (21 CFR 312.32(c)(1)(i)(A)
- One or more occurrences of an event that is not commonly associated with drug exposure, but is otherwise uncommon in the population exposed to the drug as per 21 CFR 312.32(c)(1)(i)(B)
- An aggregate analysis of specific events observed in a clinical trial (known consequences of the underlying disease or condition) that indicates those events occur more frequently in the drug treatment group than in a concurrent or historical control group as per (21 CFR 312.32(c)(1)(i)(C)
What to submit to eCTD:
- Findings from other studies as per 21 CFR 312.32(c)(1)(ii))
- Findings from animal or in vitro testing as per 21 CFR 312.32(c)(1)(iii))
- Increased rate of occurrence of serious suspected adverse reactions as per 21 CFR 312.32(c)(1)(iv))
Presubmission Consultation
The FDA encourages the sponsor to seek early consultation prior to submitting an IND safety report in electronic format either through the ESG or SRP if necessary. Presubmission consultation can avoid issuance of suggestions, mandatory changes, or clinical holds on the application and are well worth the time and effort. The FDA staff can provide valuable guidance about information necessary for an IND safety report submission.
Where To Submit IND Safety Reports?
To eCTD:
https://www.fda.gov/drugs/electronic-regulatory-submission-and-review/electronic-common-technical-document-ectd
To FAERS:
https://www.fda.gov/drugs/questions-and-answers-fdas-adverse-event-reporting-system-faers/fda-adverse-event-reporting-system-faers-electronic-submissions
Benefits Of New Process To Industry
The new guidance and process will allow FDA to receive standard data elements and be able to use data visualization and analytics tools for review and tracking in the processing and submission. For Industry, it is more comprehensive and structured format than Medwatch form, that is direct electronic submissions to FDA from the pharmacovigilance system, there is no requirement for a form 1571 or a cover letter and no need to send duplicate reports. It is also consistent with format for NDA, BLA and ex-US submissions.
References
1 https://www.ich.org
2 FDA: Providing Regulatory Submissions in Electronic Format: IND Safety Reports: Guidance for Industry (OCTOBER 2019)
3 FDA: https://open.fda.gov/data/faers/
About the Author:
Lin Zhang, M.D., senior director of a health care industry company in the United States. With the experience in clinical medicine, biotechnology, health industry and other fields, he is responsible for the research and development of plant medicine, functional food and health products. He was a clinician and worked for the National Cancer Institute, FDA and the National Cancer Center of Japan for many years.
-----------------------------------------------------------------------
Editor's Note:
To become a freelance writer of En-CPhI.CN,
welcome to send your CV and sample works to us,
Email: Julia.Zhang@ubmsinoexpo.com.




 ALL
ALL Pharma in China
Pharma in China Pharma Experts
Pharma Experts Market News
Market News Products Guide
Products Guide Brand Story
Brand Story























 Pharma Sources Insight January 2025
Pharma Sources Insight January 2025








