Protecting patients is the FDA’s highest priority, and Americans can be confident in the quality of the products the agency approves. The recent Angiotensin II Receptor Blocker (ARB) recalls have deeply concerned patients, the medical community, the agency and international regulatory bodies. The FDA is aware many patients rely on ARB medicines, and we are concerned about the presence of nitrosamine impurities in these drugs. Millions of people benefit from the high-quality products that the FDA regulates, and the U.S. has the most robust drug supply in the world, with strict standards for safety, effectiveness and quality.
Clarifying the risk and scope of exposure
As part of our efforts to be transparent regarding impurities in ARBs, we want to make sure patients have a full understanding of how these impurities may affect them. Notably, we would like to stress that the actual risk to patients is likely much lower than our estimates, which reflect a scientific assessment of the highest possible exposure. Weinitially estimated that if 8,000 people took the highest valsartan dose (320 mg) containing N-Nitrosodimethylamine (NDMA) from the recalled batches daily for four years, there may be one additional case of cancer over the lifetimes of those 8,000 people. In reality, the vast majority of patients exposed to NDMA through ARBs received much smaller amounts of the impurity than this worst-case scenario, and, since not all ARBs are affected, it’s very likely that a patient taking an ARB for four years would not have always received one of the affected products.
One question we have received from many patients and other stakeholders is how big the impact has been – how many patients have been affected? While we know the number of patients taking ARB medicines, we do not know the exact number of patients impacted by these ARB recalls. The reasons for this are nuanced. For example, we understand that health care professionals, out of an abundance of caution, may have replaced their patients’ ARB prescriptions that were not part of the announced recalls. Because patients’ prescription bottles do not always identify lot numbers, it was difficult for patients and pharmacists to determine whether a medicine was part of the recalls. Patients may have returned medication unnecessarily if their supply was not part of a recall, but they were unsure. Therefore, we estimate there were likely more drugs replaced due to recalls than necessary, which means more patients were impacted but not necessarily exposed to an impurity.
Manufacturers only estimate the number of products still in the distribution chain that are subject to recall, but not the number of impacted patients. However, through our ongoing track and trace efforts for prescription drugs in the supply chain, we’re working on ways to improve industry’s ability to track, detect and remove potentially dangerous drugs from the supply chain more rapidly and efficiently. ARB medications that contain impurities above the published interim acceptable limits continue to be recalled, with certain exceptions. However, it’s also important to underscore that there are ARB medicines that remain on the market and have been determined not to contain any nitrosamine impurities. We continue to encourage patients talk to a health care professional if they have questions about their medicine, as the risks of stopping taking an ARB product for treating high blood pressure and heart failure greatly outweighs the potential risk of exposure to trace amounts of nitrosamines.
Enhancing oversight of manufacturing data
As we continue our analysis of this situation to better understand the root causes, we’re learning more about how nitrosamine impurities may have formed and been present in ARBs. Through each step of our investigation, we’ve uncovered new information and taken a number of actions, including regulatory and advisory actions, where appropriate, to prevent the presence of unacceptable levels of these impurities. For example, earlier this month, we issued a warning letter to Lantech Pharmaceuticals Limited in Telangana, India, for current good manufacturing practice violations. Lantech acts as a contract solvent recovery facility for valsartan active pharmaceutical ingredient (API) manufacturing operations. When we inspected Lantech in March 2019, we determined that solvent recovered by the company contained N-Nitrosodiethylamine (NDEA) and that Lantech had insufficiently assessed the risks associated with their processes and did not adequately investigate the impurities. Specifically, the warning letter cites Lantech’s failure to control and monitor procedures to recover solvents to ensure that they meet appropriate standards before reuse. After we gathered and evaluated all the facts, we placed Lantech on import alert in June 2019, preventing API made using its recovered solvent from legally entering the U.S.
We continue to work closely with our global regulatory partners, including the European Medicines Agency (EMA), Health Canada, and many others, to understand the full scope of this issue. We share inspectional findings, laboratory testing methods and results, and our assessments of root cause and impact. We are working to incorporate what we have learned about the process risks that caused these impurities into our oversight of drug manufacturing, which includes how we assess applications and changes to applications, as well as enhancing our inspection coverage to evaluate the controls in place to prevent unacceptable levels of nitrosamine. For example, we plan to adjust inspections of API sites to include enhanced evaluation of impurity controls, particularly when the manufacturing process may lead to the formation of a nitrosamine or when recycled raw materials can create unacceptable contamination. In the past year, the agency has conducted multiple unannounced, for-cause inspections to evaluate the practices at various API manufacturers and to verify appropriate corrective actions to address the risk of nitrosamine contamination. We are also working to improve how companies submit manufacturing changes to the agency with the goal of further improving our review of the more than 5,000 quality-related application change requests (supplements) each year.
We continue to work with our global regulatory partners and industry as members of the International Conference on Harmonization (ICH) to publish guidance on controlling impurities in drugs and managing changes (e.g., ICHQ3A, Q3C, Q3D, Q7, Q11, and M7). These guidances and others developed through ICH are revised as new impurities and risks are identified and have helped prevent unacceptable impurities in drugs. We work with manufacturers during application review and after marketing begins over the lifecycle of their drugs to evaluate proposed process changes, conduct facility inspections, test samples of marketed drugs, evaluate complaints, and investigate issues that arise, all to protect the safety and quality of medicines.
Expanding our investigation
We have known that certain drug manufacturing processes pose a risk for forming genotoxic impurities, and this is an issue the FDA and other regulators have been working on for a number of years – well before the nitrosamine impurities were discovered in ARBs last summer. In fact, we issued guidance in early 2018 to provide information to manufacturers regarding their responsibilities to assess the risks and implement appropriate controls for their manufacturing process. Now that we know some of the root causes of the nitrosamine impurity problem, we’re using these findings to inform our evaluation of medicines other than ARBs. We are testing samples of other drugs with similar manufacturing processes. If we detect a problem, we will take appropriate action. In the meantime, we continue to work with ARB manufacturers to remove all affected drugs from the market, and we work with API manufactures to fix their processes so they do not distribute affected API to drug product manufacturers.
Our list of affected products and products without impurities remains available to the public, and we will update it when we have new information to share. We will also continue to provide timely updates about any future issues associated with this ongoing incident that may impact public health.
As we work to safeguard the quality of our medicines, the American public can expect that we will act quickly to address any issue as soon as we find out about it to prevent as much harm to patients as possible. We’re also committed to communicating as transparently as possible. When we found out about the impurities in ARB products, we worked with industry and our international regulatory colleagues to understand the situation and take preventative measures. We have also communicated frequently over the past year to share timely safety and manufacturing information. We have consistently and transparently communicated with pharmacists, patients and health care professionals about the ARB recalls as they have evolved.
Ultimately, our goal is to be certain that no ARBs with unacceptable impurity levels reach patients. Based on our current assessments, including lab testing, the agency has identified 43 ARB medications that have been determined not to contain any nitrosamine impurities. As we continue our assessments and as companies continue to manufacture ARBs without nitrosamine impurities to replenish the U.S. supply, we expect this figure to rise. We know that ARBs can be produced without nitrosamine impurities, and we are working with manufacturers to reach that point.
The FDA has ongoing review, surveillance, compliance and pharmaceutical quality efforts across every product area, and we will continue to work with drug manufacturers to ensure safe, effective, and quality drug products for the American public.
-----------------------------------------------------------------------
Editor's Note:
For any copyright disputes involving the content,
please email: Julia.Zhang@ubmsinoexpo.com to delete.




 ALL
ALL Pharma in China
Pharma in China Pharma Experts
Pharma Experts Market News
Market News Products Guide
Products Guide Brand Story
Brand Story


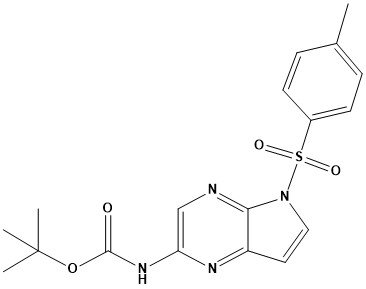
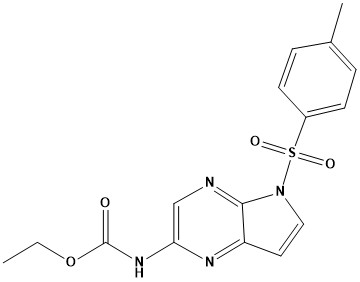
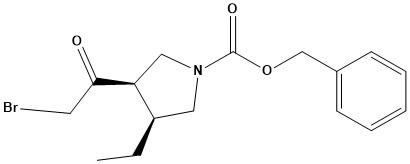
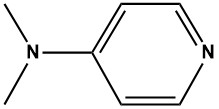


















![N - alpha [(h - fluorene 9-9 - methoxy) carbonyl] - N epsilon - [(4 - methoxy phenyl) diphenyl methy](https://eimg.pharmasources.com/img_Cphi_en/Product/2018_06/Mimg_1806191042684010.jpg)







 Pharma Sources Insight January 2025
Pharma Sources Insight January 2025








