The U.S. Food and Drug Administration (FDA) cleared for marketing four previously cleared tests with new indications to aid in the diagnosis of Lyme disease. The tests cleared today are the first time that a test has been indicated to follow a new testing paradigm in which two tests called enzyme immunoassays (EIA) are run concurrently or sequentially, rather than the current two-step process in which a separate protein test called a Western Blot must be run after the initial EIA test.

"Lyme disease can have a devastating impact on patients. With today's action, clinicians have a new option to test for Lyme that is easier to interpret by a clinical laboratory due to the streamlined method of conducting the test. These tests may improve confidence in diagnosing a patient for a condition that requires the earliest possible treatment to ensure the best outcome for patients," said Tim Stenzel, M.D., Ph.D., director of the Office of In Vitro Diagnostics and Radiological Health in the FDA's Center for Devices and Radiological Health.
Lyme disease is caused by the bacteria Borrelia burgdorferi and is transmitted to humans through the bite of infected ticks. Typical symptoms include fever, headache, fatigue and skin rash called erythema migrans. If left untreated, infection can spread to joints, the heart and the nervous system. In 2017, the last year for which the Centers for Disease Control and Prevention (CDC) has published data, a total of 42,743 confirmed and probable cases of Lyme disease were reported to CDC, an increase of 17% from 2016.
Laboratory diagnosis of Lyme disease has traditionally used a two-tier process for detecting the presence of antibodies against Borrelia burgdorferi in a patient's blood. Antibodies are proteins present in the blood when the body is responding to a specific infection. In the previous two-tier approach, different types of tests were used (EIA and Western blots) to confirm a clinical diagnosis. The tests cleared today involve a modified approach that uses only EIA technology-based tests.
The FDA reviewed data from clinical studies of the ZEUS ELISA Borrelia VlsE1/pepC10 IgG/IgM Test System, ZEUS ELISA Borrelia burgdorferi IgG/IgM Test System, ZEUS ELISA Borrelia burgdorferi IgM Test System, and the ZEUS ELISA Borrelia burgdorferi IgG Test System that showed this alternative approach, referred to as a modified two-tier test, is as accurate as current methods for detecting antibodies for assessing exposure to Borrelia burgdorferi, the causative agent of Lyme disease, over current methods.
CDC recommendations should be followed for the diagnosis of Lyme disease and for determining when laboratory tests are appropriate.
The enzyme immunoassay tests were reviewed through the premarket notification (510(k)) pathway. A 510(k) is a premarket submission made to the FDA to demonstrate that the device to be marketed is at least as safe and effective, that is, substantially equivalent, to a legally marketed device.
The FDA granted clearance of the ZEUS ELISA enzyme immunoassay tests to ZEUS Scientific.
-----------------------------------------------------------------------
Editor's Note:
If you have any suggestion to the content,
please email: Julia.Zhang@ubmsinoexpo.com




 ALL
ALL Pharma in China
Pharma in China Pharma Experts
Pharma Experts Market News
Market News Products Guide
Products Guide Brand Story
Brand Story


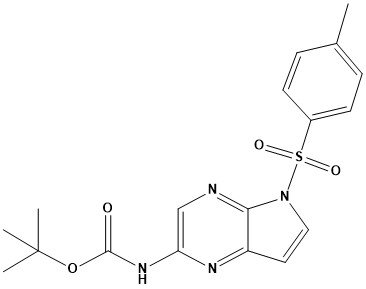
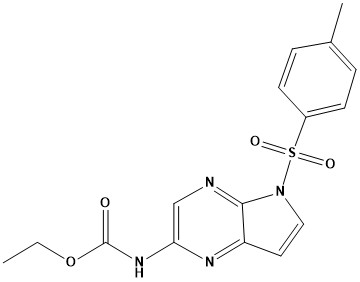
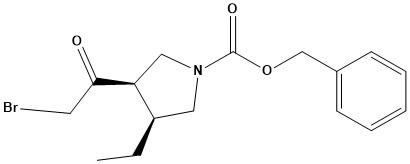
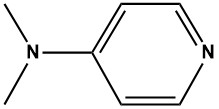


























 Pharma Sources Insight January 2025
Pharma Sources Insight January 2025








