The FDA has set itself a new goal: to become the international leader in medical device safety through better use of big data.
The agency pledged to consistently place first among the world’s regulatory agencies in identifying and reacting to device safety issues by transitioning to an active surveillance system built on real-world evidence and by seeking new authority to take faster actions for postmarket safety.
The effort also includes plans for modernizations of the 510(k) review process—with details to be released next week—as well as targeted measures related to devices for women’s health, such as surgical mesh for transvaginal repair.
"We’re evolving beyond our current post-market surveillance system—which is largely passive and relies on device users to report problems to us, sometimes resulting in underreporting," FDA Commissioner Scott Gottlieb and CDRH Director Jeff Shuren said in an agency statement outlining the first new steps of the plan.
"We’re moving to an active surveillance system that relies on real-world evidence and timely receipt of robust safety information," they wrote. "We have long recognized the systemic weaknesses of the passive system—a challenge faced by other countries—and we prioritized this area for regulatory reform efforts."
The moves build upon the FDA’s unique device identification initiative, which tags each device with a code that can be tracked across its distribution and use in patients. Its public database now contains more than 1.5 million records, the agency said, to help it link adverse event reports with specific devices.
"We believe that including the device identifier in electronic data more broadly, including in insurance claims, will advance FDA’s efforts to leverage real-world data to support the development of more effective post-market surveillance tools," Gottlieb and Shuren wrote.
In addition, the agency is committing new resources to rapidly enable its National Evaluation System for health Technology, or NEST, for the analysis of real-world data from patient registries, Medicare claims and electronic medical records.
NEST will also provide information to medical device manufacturers developing improvements for their devices, the agency said.
Its independent governing board is run from a public-private coordinating center, dubbed NESTcc, that is comprised of industry and community stakeholders as well as patient and provider representatives. FDA provided the seed funding for NESTcc and secured partial industry funding through the latest iteration of the agency’s user fee legislation.
NESTcc has entered agreements with 12 organizations—representing more than 195 hospitals and nearly 4,000 outpatient clinics with access to more than 495 million patient records—to help build out its data network.
The agency allocated $3 million to NESTcc in September, in addition to the annual funding allocated by the latest user fee agreement. The Trump administration has also requested an additional $46 million for fiscal 2019 to support NEST.
Earlier this month, NESTcc announced eight real-world data demonstration projects, exploring the use of patient registry and claim data in evaluating total joint replacement and spine fusion surgeries, as well as the effectiveness of different tissue closure techniques following surgery.
In women’s health, the FDA has scheduled an advisory committee meeting for February 2019 to discuss surgical mesh intended for transvaginal pelvic organ prolapse repair and to consider additional regulatory actions. Two years ago, the FDA reclassified surgical mesh devices from Class II to Class III, requiring manufacturers to submit them for premarket approval and leaving only three products on the market for the indication.
The agency also pointed to its National Breast Implant Registry launched in September following public concerns that implants may be linked to health conditions such as chronic fatigue, cognitive issues and muscle pain, though the FDA has not yet found evidence of a direct association.
The agency is also continuing to monitor energy-based devices which may be inappropriately marketed for vaginal rejuvenation and symptoms related to menopause, urinary incontinence or sexual function. This past summer, the FDA publicly warned seven manufacturers of its concerns—since then, all seven have made changes to their websites to remove those unapproved claims.




 ALL
ALL Pharma in China
Pharma in China Pharma Experts
Pharma Experts Market News
Market News Products Guide
Products Guide Brand Story
Brand Story


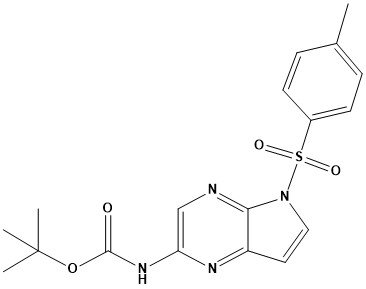
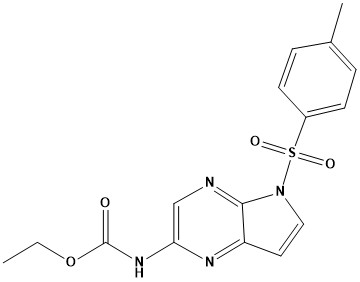
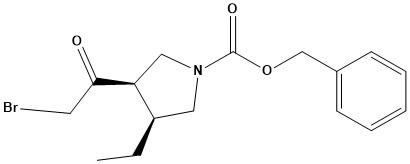
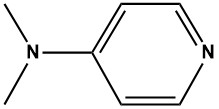


























 Pharma Sources Insight January 2025
Pharma Sources Insight January 2025








