An abnormal form of the tau protein that aggregates in neurofibrillary tangles has long been implicated in Alzheimer’s disease, though the exact mechanisms behind tau's role in the disease are unknown. Now a team of scientists led by Massachusetts General Hospital and Johns Hopkins suggest that the protein disrupts signaling within neurons to cause the disease.
By studying postmortem human Alzheimer’s brain tissue and in vitro and mouse models, the researchers found that pathological tau impairs the nuclear pore complex (NPC), a key part of the nuclear membrane that acts as the gateway for the exchange of proteins and RNA with the surrounding cytoplasm. In addition, defects in these pores might further lead to tau accumulation inside neurons. The team described their findings in the journal Neuron.
Small molecules can pass freely through the NPC, but larger molecules require assistance from interactions between transport receptors and proteins called nucleoporins, or nups, which form NPC. A signaling protein called Ran is necessary for the nuclear transport, as it regulates whether the molecule is imported or exported through the NPC.
Previous studies have linked disrupted nucleocytoplasmic transport in neurons with several neurodegenerative diseases, including amyotrophic lateral sclerosis, frontotemporal dementia and Huntington’s, as well as normal human aging. "In other systems, disruption of this communication causes cell misfunction and even cell death," said co-senior author Bradley Hyman of Massachusetts General Hospital, in a statement.
Hyman and his colleagues wanted to test their hypothesis that a similar phenomenon might be happening in Alzheimer's. They first experimented on neurons from patients with Alzheimer’s and on cellular models, and they found that Alzheimer’s-associated phosophorylated tau directly interacts with one of the most abundant nucleoporins, called Nup98. Instead of being evenly distributed in the nuclear membrane, as seen in normal control panels, the Nup98 pores were disrupted in the presence of tau, as they appeared to be depleted, and they coalesced with each other.
What’s more, the team noted that Nup98 seemed to leak into the cytoplasm, where it promoted tau aggregation. The more severe the disease was when patients were alive, the worse the Nup98 mis-location was, the team found.
They also discovered NPC leakage in mice that were genetically modified to over-express tau and develop neurofibrillary tangles. The rodents showed Ran depletion from the nucleus that appeared to correlate with the misplacement of Nups into the cytoplasm. Interestingly, after scientists suppressed the expression of abnormal tau, levels of both Ran and Nup98 in the NPC were restored.
While defective tau has long been associated with Alzheimer’s, recent research efforts have instead focused on tackling beta-amyloid, which is also widely believed to cause the condition. However, clinical studies in that field are filled with frustrations. An amyloid-fighting antibody from Biogen and Eisai recently showed cognitive benefits but still sparked debates over the significance of the data, as well as the whole amyloid theory.
The Massachusetts General Hospital and Johns Hopkins researchers suggest in their new study that preventing tau interactions with Nup98 may be promising in treating Alzheimer’s and other neurodegenerative diseases. "One of the exciting things about these findings is that, if we can block the interaction between tau and the nuclear pore, it might allow existing neurons to become more functional in patients," Hyman said.
-----------------------------------------------------------------------------
Editor's Note:
To apply for becoming a contributor of En-CPhI.cn,
welcome to send your CV and sample works to us,
Email: Julia.Zhang@ubmsinoexpo.com.




 ALL
ALL Pharma in China
Pharma in China Pharma Experts
Pharma Experts Market News
Market News Products Guide
Products Guide Brand Story
Brand Story


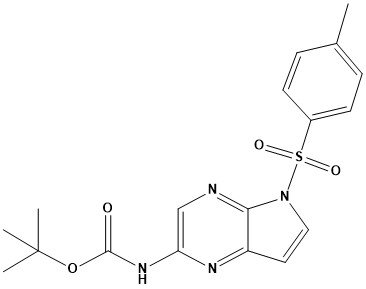
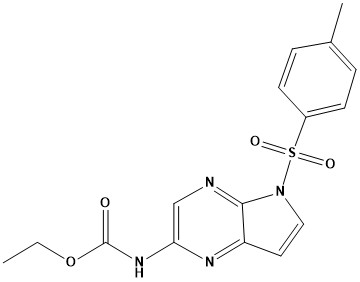
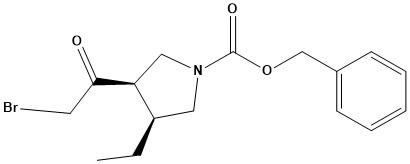
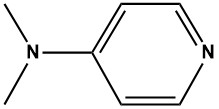

















 Pharma Sources Insight January 2025
Pharma Sources Insight January 2025








