Foundation Medicine, Inc. (NASDAQ:FMI) today announced that the U.S. Food and Drug Administration (FDA) granted a Breakthrough Device designation (formerly Expedited Access Pathway program) for its new liquid biopsy assay, which is an expanded version of its FoundationACT® assay. The new assay will include more than 70 genes and genomic biomarkers for microsatellite instability (MSI) and blood tumor mutational burden (bTMB). If approved, this test could be the first FDA-approved liquid biopsy assay to incorporate multiple companion diagnostics (CDx) and multiple biomarkers to inform the use of targeted oncology therapies, including immunotherapies.
"Liquid biopsies are becoming an increasingly important option to inform personalized treatment decisions for patients with advanced cancer. A critical need exists for non-invasive solutions for these patients to help guide the use of both targeted and immunotherapies. After successfully completing the parallel review process with FoundationOne CDx™ for solid tumors, we’re pleased to continue our work with the FDA applying this expertise to our liquid biopsy franchise with the potential to offer oncologists multiple FDA-approved options that enable biomarker-driven care and ultimately better outcomes for patients," said Vincent Miller, M.D., chief medical officer at Foundation Medicine. "The acceptance of this assay for Breakthrough Device designation is an important step to advancing precision medicine options for patients, including the potential intended use as a companion diagnostic across multiple types of cancer, which will also help our biopharma partners accelerate their development efforts for these programs."
The new liquid biopsy assay is a next generation sequencing-based in-vitro diagnostic device for the detection of substitutions, insertion and deletion alterations (indels), copy number alterations (CNAs) and select gene rearrangements, as well as genomic signatures including microsatellite instability (MSI) and blood tumor mutational burden (bTMB) using circulating cell-free DNA (cfDNA) isolated from plasma derived from peripheral whole blood. The company plans to seek approval of the assay for use as a companion diagnostic to identify patients who may benefit from treatment with certain targeted therapies in accordance with the approved therapeutic product labeling and to provide tumor mutation profiling to be used by qualified health care professionals following the professional guidelines in oncology for patients with malignant neoplasms.
The FDA granted Foundation Medicine's request for Breakthrough Device designation because it met the criteria necessary for inclusion in the program, one of which is the large unmet medical need for comprehensive genomic profiling of tumors for cancer patients for whom a tissue sample is unavailable for testing. Under the Breakthrough Device Program, the FDA works with device sponsors to try to reduce the time and cost from development to an approval decision.
About Foundation Medicine
Foundation Medicine (NASDAQ:FMI) is a molecular information company dedicated to a transformation in cancer care in which treatment is informed by a deep understanding of the genomic changes that contribute to each patient's unique cancer. The company offers a full suite of comprehensive genomic profiling assays to identify the molecular alterations in a patient's cancer and match them with relevant targeted therapies, immunotherapies and clinical trials. Foundation Medicine’s molecular information platform aims to improve day-to-day care for patients by serving the needs of clinicians, academic researchers and drug developers to help advance the science of molecular medicine in cancer.
Foundation Medicine® and FoundationACT® are registered trademarks and FoundationOne CDx™ is a trademark of Foundation Medicine, Inc.
Cautionary Note Regarding Forward-Looking Statements for Foundation Medicine
This press release contains "forward-looking statements" within the meaning of the Private Securities Litigation Reform Act of 1995, including, but not limited to, statements regarding the technical specifications of the company’s new liquid biopsy assay; the scope and timing of any approval of the liquid biopsy assay as a medical device by the FDA, including its potential approval and use as a companion diagnostic; the liquid biopsy assay’s ability to inform the use of targeted oncology therapies, including immunotherapies, or enhance patient access to targeted therapies and clinical trials; and any benefits provided by an FDA-approved version of the liquid biopsy assay, including benefits to patients, physicians or biopharma partners. All such forward-looking statements are based on management's current expectations of future events and are subject to a number of risks and uncertainties that could cause actual results to differ materially and adversely from those set forth in or implied by such forward-looking statements. These risks and uncertainties include the risks that the FDA does not approve the company’s new liquid biopsy assay as a medical device; the FDA is delayed in the completion of its review process; the liquid biopsy assay does not accelerate the development efforts of the company’s biopharma partners; and the risks described under the caption "Risk Factors" in Foundation Medicine's Annual Report on Form 10-K for the year ended December 31, 2017, which is on file with the Securities and Exchange Commission, as well as other risks detailed in subsequent filings with the Securities and Exchange Commission. All information in this press release is as of the date of the release, and Foundation Medicine undertakes no duty to update this information unless required by law.
Register as Visitor to CPhI China 2018!




 ALL
ALL Pharma in China
Pharma in China Pharma Experts
Pharma Experts Market News
Market News Products Guide
Products Guide Brand Story
Brand Story


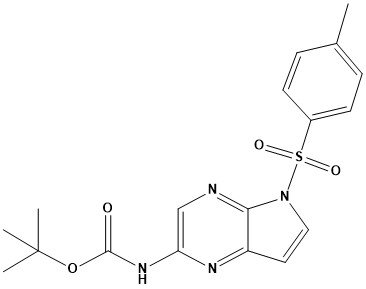
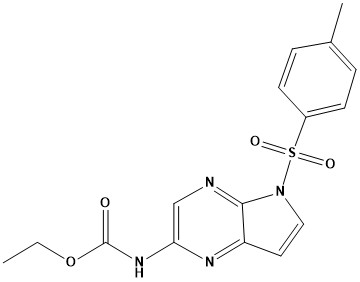
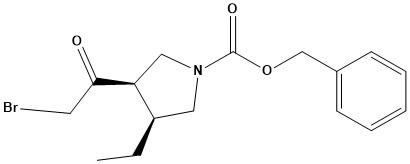
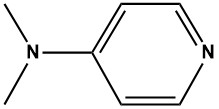


























 Pharma Sources Insight January 2025
Pharma Sources Insight January 2025








