It gradually approaches the Chinese New Year holidays, but CFDA has never stopped its pace of intending to rectify the industry and continuing to introduce new policies. The Center for Drug Evaluation (CDE) under CFDA has introduced a draft for comment of new guidelines, i.e., Technical Guidelines for the Process Change Study of Marketed Chemical Pharmaceutical Products. This draft for comment has been hotly discussed by the industry upon introduction. There are different guesses in the industry: someone guessed this work to be the foreshadowing and preparation for the process checking that was not completed in 2016; someone guessed that introduction of the guidelines was to regulate the chemical drug process change work and promote the normative proceeding of the supplementary applications.
I explain the document as follows according to my own understanding:
Firstly, this document is very important
The reason for this is that, in the long lifecycle management, any pharmaceutical product process or even prescription may be changed with changes of external conditions or driving factors for internal improvement. So to speak, changing is eternal and unchanging is false. The inconsistency between registered process and actual process of many enterprises is caused by the previous unscientific registered process change management.
Secondly, the time of introduction to this document shall be thought over
It has been 8 years since the introduction of the last relevant document Technical Guidelines for the Change Study of Marketed Chemical Pharmaceutical Products (I) in 2008, which is supposed to be revised and updated, but pharmaceutical industry people would find that the introduction to the new draft for comment is expected to cooperate with the late-stage process checking, in combination with the registered process checking issue introduced in 2016 but set aside at present. This guess is not groundless.
Thirdly, this document shows some progress
We can see that the author of this document put much thought into it, from its content and length. And the references of the document list Chinese and foreign guidelines or regulations relating to pharmaceutical product process change, for example, in the preparation process change part, it mentions for many times that the new process or new prescription shall be compared with the original pharmaceutical products.
Fourthly, the defects and shortcomings of this document are obvious
Next, I would like to talk about the shortcomings of this document according to my understanding of the pharmaceutical industry.
Defect 1: The view is inappropriate that pharmaceutical product manufacturing enterprises are the subject of the production process change study and study result self-assessment.
This view is proposed based on the old pharmaceutical product registration system and is mentioned in 2008 edition, but it is inappropriate in 2017, because the MAH System has been started the pilot work in 10 provinces in 2016 and according to requirements of MAH System, natural persons or pharmaceutical research institutions can be the holders of pharmaceutical product production approval, which is different from the previous requirement that the pharmaceutical product production approval must be held by manufacturing enterprises.
Defect 2: Many requirements are inconsistent.
For example, regarding the stability study requirement, the front of the document mentions requiring 3-6 months’ accelerated test and long-term test, but this time changes to 6 months’ test later in the document.
Defect 3: There is no terms part, but new terms are made at will.
The document has a defect that there are no definitions of noun terms, but new terms are made at will in the text. For example, special excipients are mentioned in the excipients requirement part, but what are the special excipients? There is no explanation.
Defect 4: Mix up studies of different R&D phases
For example, the preparation prescription screening part mentions that stress test should be conducted, which is completely wrong. To tell the truth, this problem is not made clear by many research persons, either.
The following table can help understand the differences between compatibility test and stress testing.
|
Term
|
Applicable object
|
R&D phase
|
Test condition
|
Purpose and significance
|
|
Compatibility test
|
Only preparation products
|
Pre-prescription phase, for screening the best prescription
|
The typical conditions are high humidity, high temperature and lighting
|
For determining if the raw materials and excipients are compatible.
|
|
Stress test
|
Preparation and API products
|
Generally selecting pilot test phase samples for proceeding
|
For preparations: Generally, high humidity, high temperature and lighting conditions;
For APIs: Besides high humidity, high temperature and lighting conditions, possibly many other conditions like acid and alkali damage
|
It is the trial test of the stability test, for choosing packaging materials, temperatures for accelerated and long-term tests, and for determining the indicated analytical methods.
|
In short, this document has less than 70 pages while the 2008 guidelines have 82 pages. There are not many highlights in this document. According to my overall feeling, the author does not have deep overall understanding of R&D with insufficient multidisciplinary knowledge preparation. CDE is recommended to revise this draft for comment carefully.




 ALL
ALL Pharma in China
Pharma in China Pharma Experts
Pharma Experts Market News
Market News Products Guide
Products Guide Brand Story
Brand Story

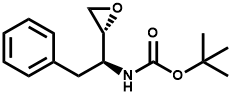
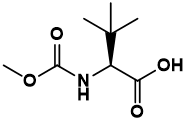

















 Pharma Sources Insight January 2025
Pharma Sources Insight January 2025








